3D binding pocket protein structure similarity virtual screening
|
 |
|
|
|
|
|
|
|
|
|
|
|
|
|
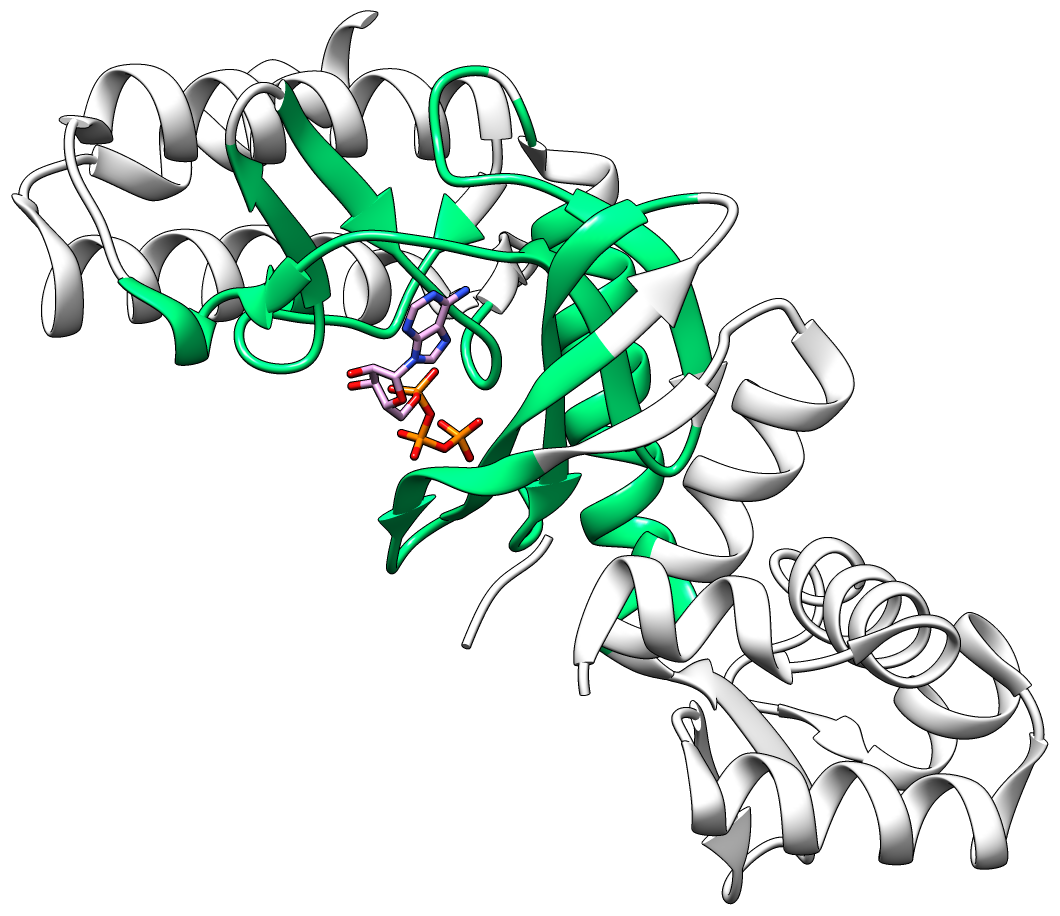
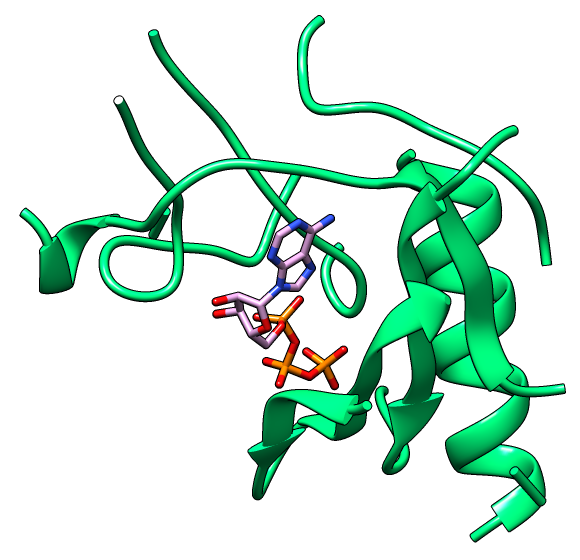
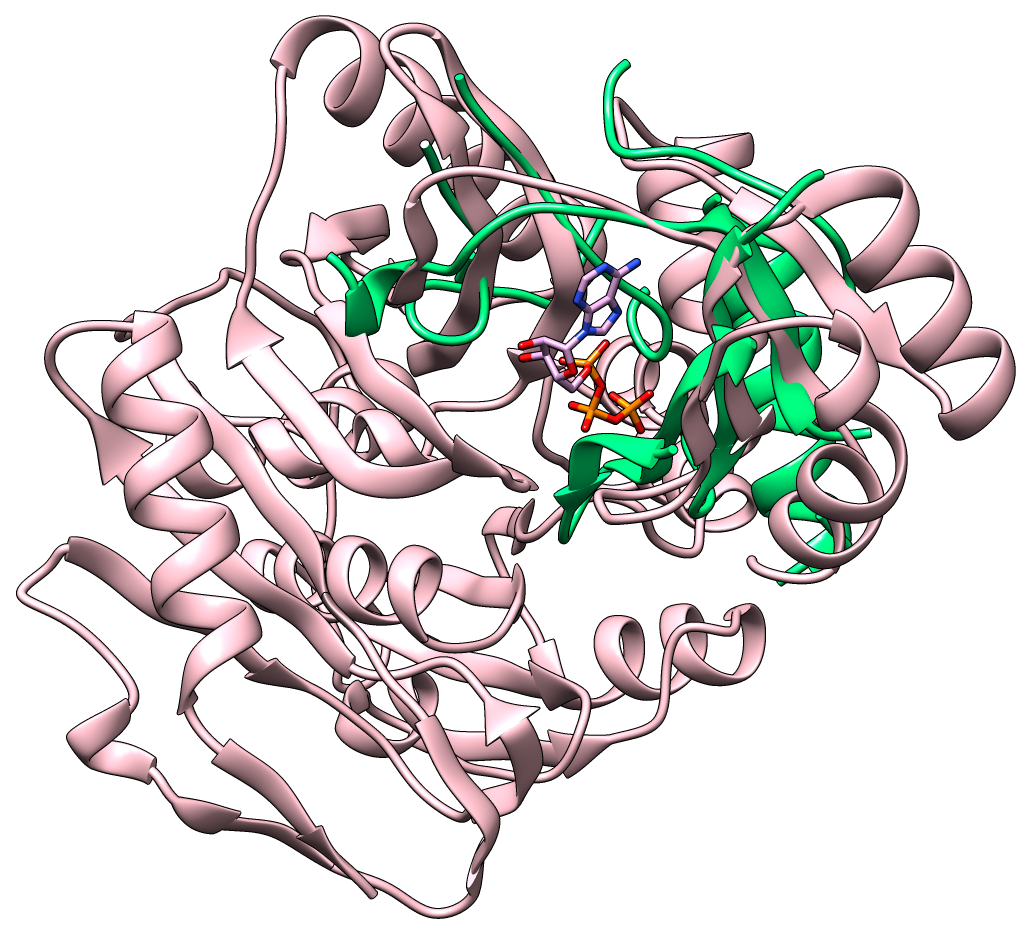
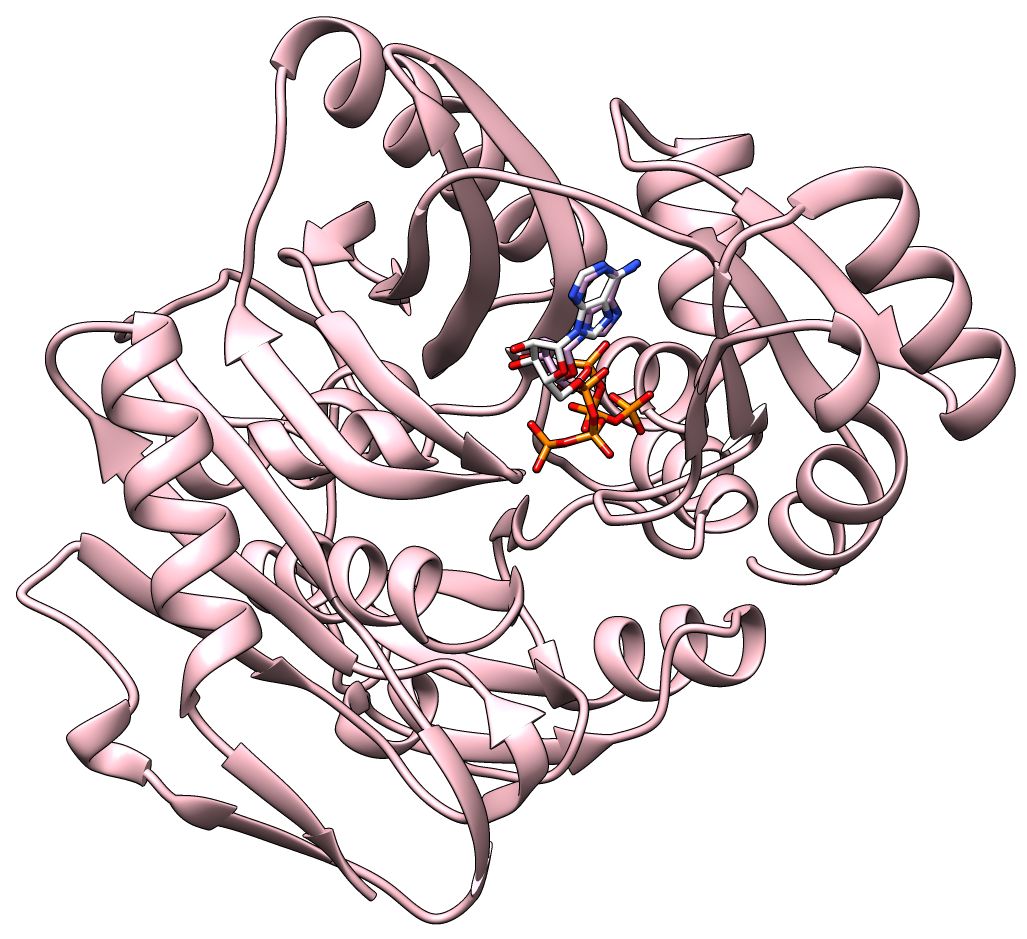
Current drug discovery efforts usually identify one or a few protein target(s) and attempt to inhibit these using small molecules drugs. Most inhibitors/drugs are identified using various computational/experimental screens followed by rounds of rational manipulation and extensive experimental validation. The drugs bind selectively to a pocket on the targets because of complementarity in shape and physicochemical properties. However, the diversity of protein shapes is limited, and it is likely that similar binding pockets could be found in other proteins. The binding of the drug to these off-target proteins could either lead to adverse drug reactions or indicate an alternate use of the drug. An efficient drug discovery effort would be strengthened with the identification of putative binding pockets on off-target proteins. We have developed CLICK algorithm (Nguyen and Madhusudhan, Nucleic Acids Res., 2011) and web server (Nguyen et al., Nucleic Acids Res., 2011) for comparing a pair of 3D structures of molecules. CLICK creates small cliques of points from representative atoms of spatially proximal amino acid residues (3−7 in number). These cliques are then superimposed by a 3D least-squares fit. To guide the matching of cliques, other features such as solvent accessibility, secondary structure, and residue depth can also be used. CLICK is capable of aligning structures with dissimilar topologies, conformations, or even molecular types. These unique properties make CLICK particularly well suited for comparing protein substructures, such as ligand binding sites. In addition, CLICK has been extensively benchmarked to other popular methods for protein structural alignments and comparison of binding sites (Nguyen and Madhusudhan, Nucleic Acids Res., 2011, Brown et al., Bioinformatics, 2015, Garma et al., Proteins, 2016, and Zhang and Pyle, iScience, 2022). In this study, we have developed and optimized CLICK algorithm to make a significant enhancement in the capability of CLICK to include virtual screening for similar ligand binding sites over entire databases and identification of potential target proteins of ligands/small molecules . We are firmly of the belief that our enhanced web server (3Dclick) offers researchers an opportunity for virtual screening different databases including AlphaFold structures at EBI (https://alphafold.ebi.ac.uk/) for similar protein-ligand binding sites. All of this should not only enable identification of potential target proteins of ligands/small molecules but also aid in the general area of protein/inhibitor design. The below Figure 1A shows the 3D structure of A.fulgidus Rio2 Kinase (gray ribbons) complexed with ATP (Adenosine triphosphate, as shown in sticks) (PDB (Protein Data Bank) code: 1zaoA, Laronde-Leblanc et al., FEBS J , 2005). The binding pocket/site of A.fulgidus Rio2 Kinase with ATP is displayed in green ribbons. Figure 1B shows the 3D structure of binding site of A.fulgidus Rio2 Kinase (green ribbons) with ATP (sticks), which is used for structure similarity virtual screening with 3Dclick. As shown in Figure 1C, PurT-encoded glycinamide ribonucleotide transformylase (pink ribbons) (PDB code: 1kj9A, Thoden et al., J Biol Chem , 2002) is identified as a target protein of ATP using 3Dclick that has similarity scores with the ATP binding site of A.fulgidus Rio2 Kinase (Z-score = 2.98; Structure Overlap = 67.39%; RMSD = 2.06 Å). Similarly to CLICK, 3Dclick produces a Z-score for reliability of match, and we had previously established that a score of 2 and above was indicative of a significant comparison of 3D structure/substructures (Nguyen et al., J. Chem. Inf. Model., 2019). Interestingly, A.fulgidus Rio2 Kinase and PurT-encoded glycinamide ribonucleotide transformylase are unrelated by protein fold and sequence. The Structural Classification of Proteins (SCOP) of A.fulgidus Rio2 Kinase are a.4.5.56 and d.144.1.9 while SCOP of PurT-encoded glycinamide ribonucleotide transformylase are c.30.1.1, d.142.1.2, and b.84.2.1. Figure 1D shows the possible complex structure of PurT-encoded glycinamide ribonucleotide transformylase (pink ribbons) and ATP (plum and orange sticks) using 3Dclick superimposition of binding site of A.fulgidus Rio2 Kinase with ATP (Figure 1B) on the structure of PurT-encoded glycinamide ribonucleotide transformylase (Figure 1C). As indicated in Figure 1D, the ATP in X-ray structure of PurT-encoded glycinamide ribonucleotide transformylase (white and orange sticks) has the same location as ATP predicted by 3Dclick (plum and orange sticks). This result indicates that 3Dclick has capability of predicting potential target proteins of ligands/small molecules even for unrelated protein folds with the query ligand binding site. Several web servers have been developed for predicting potential target proteins of small molecules. idTarget web server (Wang et al., Nucleic Acids Res., 2012) predicts possible binding target proteins of a small chemical molecule via a divide-and-conquer docking approach and has been shown to be able to reproduce known (off-)targets of drugs or drug-like compounds. A recent study has shown that CLICK identified potential human target proteins for the anticancer drug Nutlin with better metrics than those from idTarget (Nguyen et al., J. Chem. Inf. Model., 2019). ProBiS-ligands web server (Konc and Janežič, Nucleic Acids Res., 2014) identifies target proteins in the PDB that have similar structures with the binding site of a query structure using the ProBiS algorithm (Konc and Janežič, Bioinformatics, 2010) for detection of structurally similar protein binding sites by local structural alignment. The current limitation of ProBiS-ligands is only to find target proteins in the PDB. To the best of our knowledge, GrAfSS (Ghani et al., Nucleic Acids Res., 2022) and our 3Dclick are the first web servers for predicting potential target proteins of small molecules/drugs on the database of AlphaFold structures. However, GrAfSS web server only identifies target proteins for the ligand binding sites or substructure with a maximum of 12 residues. In addition, both ProBiS-ligands and GrAfSS web servers could not find PurT-encoded glycinamide ribonucleotide transformylase (PDB code: 1kj9A, Figure 1C) as a target protein for ATP from the binding site of A.fulgidus Rio2 Kinase (PDB code: 1zaoA, Figure 1B) while idTarget web server is currently unreachable. | |
 |
 |
A |
B |
 |
 |
C |
D |
|
Figure 1A shows the 3D structure of A.fulgidus Rio2 Kinase (gray ribbons) complexed with ATP (Adenosine triphosphate, sticks) (PDB code: 1zaoA, Laronde-Leblanc et al., FEBS J , 2005). The binding pocket/site of A.fulgidus Rio2 Kinase with ATP is displayed in green ribbons. Figure 1B shows the 3D structure of binding site of A.fulgidus Rio2 Kinase (green ribbons) with ATP (sticks), which is used for structure similarity virtual screening with 3Dclick. The binding site of A.fulgidus Rio2 Kinase with ATP is identified using the distance cutoff of 12 Å, i.e. for each binding residue, there is at least one non-hydrogen atom of the binding residue that its distance to at least one non-hydrogen atom of ATP is less than 12 Å. The cutoff of 12 Å of binding residues is selected as in our previous study for target proteins of diacetyl, this cutoff was used to identify significant hits that agree with experimental technique of isothermal dose response-cellular thermal shift assays (CETSA) (Jafari et al., Nature Protocols , 2014). As shown in Figure 1C, 3Dclick identified PurT-encoded glycinamide ribonucleotide transformylase (pink ribbons) (PDB code: 1kj9A, Thoden et al., J Biol Chem , 2002) as a target protein of ATP with similarity scores with the ATP binding site of A.fulgidus Rio2 Kinase (Z-score = 2.98; Structure Overlap = 67.39%; RMSD = 2.06 Å). Z-score of 2 and above is indicative of a significant comparison of 3D structure/substructures (Nguyen et al., J. Chem. Inf. Model., 2019). Surprisingly, A.fulgidus Rio2 Kinase and PurT-encoded glycinamide ribonucleotide transformylase are unrelated by protein fold and sequence. Figure 1D shows the possible complex structure of PurT-encoded glycinamide ribonucleotide transformylase (pink ribbons) and ATP (plum and orange sticks) using 3Dclick. As shown in Figure 1D, the ATP in X-ray structure of PurT-encoded glycinamide ribonucleotide transformylase (white and orange sticks) has the same location as ATP predicted by 3Dclick (plum and orange sticks). | |