3D binding pocket protein structure similarity virtual screening
|
 |
|
|
|
|
|
|
|
|
|
|
|
|
Algorithm |
|
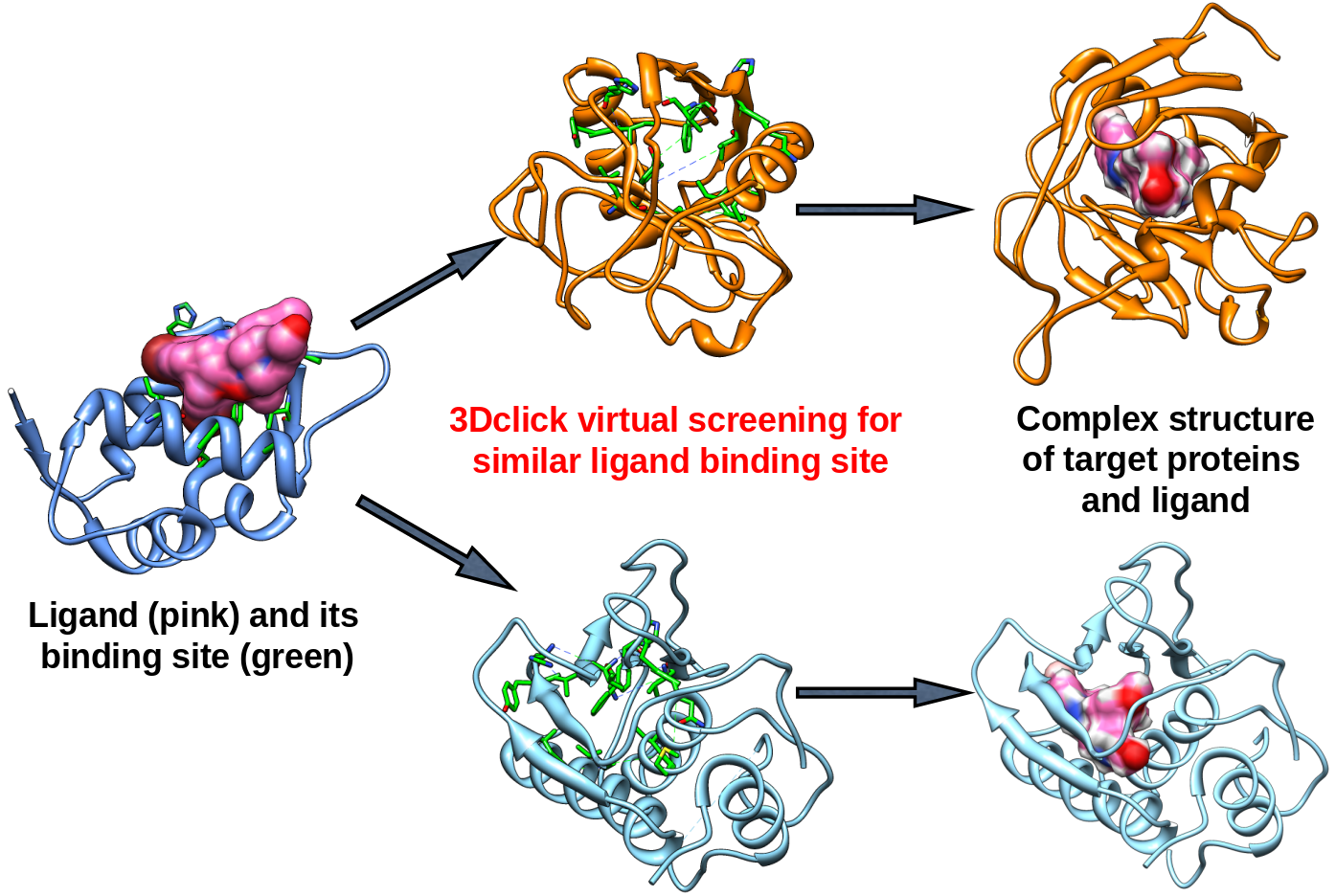
1. 3Dclick for virtual screening of similar protein-ligand binding sites
|
|
|
Figure 1: 3Dclick for virtual screening of similar protein-ligand binding sites on different databases. |
|
|
|
In our 3Dclick web server, we have developed and optimized CLICK algorithm (Nguyen and Madhusudhan, Nucleic Acids Res., 2011) to made a significant enhancement in the capability of CLICK to include virtual screening for similar protein-ligand binding sites on different databases (Small SCOP fold representatives, SCOP families (2868 PDB chains), Proteins (32,647 PDB chains NR 95), E.coli AlphaFold (4363 structures), S.cerevisiae AlphaFold (6039 structures), Human AlphaFold (23,391 structures), and User-uploaded database) as well as identification of potential target proteins of ligands/small molecules. As shown in Figure 1, 3Dclick first extracts protein-ligand binding residues from the query protein structure and ligand. The binding site/residues of protein with ligand is identified using the distance cutoff of 12 Å, i.e, for each binding residue, there is at least one non-hydrogen atom of the binding residue that its distance to at least one non-hydrogen atom of ligand is less than 12 Å. The cutoff of 12 Å of binding residues is selected as in our previous study for target proteins of diacetyl, this cutoff was used to identify significant hits that agree with experimental technique of isothermal dose response-cellular thermal shift assays (CETSA) (Jafari et al., Nature Protocols , 2014). The binding site of ligand is then used for structure similarity virtual screening on the database (Figure 1). Instead of using the main chain atoms Cα as representative atoms in CLICK (Figure 2), 3Dclick chooses the side chain Cβ atoms of the binding residues as representative atoms for structural superimposition. A clique of points is made with the representative Cβ atoms such that no pair of atoms within a clique is separated by more than a distance threshold of 10 Å. The clique size is optimized to contain 7 residues. Also, amino acid residue secondary-structure state and side chain solvent accessible area of binding residues determine what pair of cliques are matched. In addition, since binding sites of the same ligand on different proteins usually have conserved residues we have optimized CLICK algorithm to prioritize matching of conserved residues in the clique matching step for 3Dclick. We have aslo improved the running time of 3Dclick, and therefore 3Dclick performs comparisons of 3D protein structures/substructures significantly faster compared to those of CLICK. For instance, the running times for the comparison of 3D protein structures between the binding site of A.fulgidus Rio2 Kinase with ATP (PDB code: 1zaoA with 92 residues) and human AlphaFold structure of Obscurin (AF-Q5VST9-F32 with 1400 residues) are 2 seconds for 3Dclick and 15 seconds for CLICK. Moreover, in order to reduce overall run time for virtual screening on the database, we have developed and run 3Dclick in parallel on our web server. In the virtual screening step (Figure 1), 3Dclick superimposes the proteins from the database onto the ligand binding sites. Proteins that have regions that match the ligand binding sites with significant Z-scores (Z-score ≥2) are termed as “hits” or possible target proteins. The ligand is then independently transferred as rigid body onto the target protein to form a complex as shown in Figure 1. Steric hindrances in the complex and interactions between ligand and target protein are quantified by numbers of good contacts, bad contacts, and ugly contacts that are defined using the contact criteria of Maestro Schrödinger software, i.e, Contact score beween an atom of ligand and an atom of target protein is defined as: |

|
|
Here, a contact beween two non-hydrogen atoms of ligand and target protein is defined as a good contact if their Contact score >0.89 and ≤1.30, bad contact if Contact score >0.75 and ≤0.89, and ugly contact if Contact score ≤0.75. Ideally, we would want no ugly contacts between the non-hydrogen atoms of target proteins and the ligand in the complexes. Hence, 3Dclick first eliminates proteins that have the number of ugly contacts ≥ 2.2*(the number non-hydrogen atoms of ligand) from the list of possible target proteins. The energy minimization step is then applied for the complexes of target proteins and the ligand to attempt removing any possible ugly and bad contacts. After the energy minimization step, 3Dclick eliminates any proteins in the remaining list of possible targets that still have ugly contacts with the ligand. |
|
|
|
2. CLICK algorithm for comparison of 3D protein structures/substructures
|

|
|
|
|
Figure 2: A pictorial representation of the matching of n-body cliques in CLICK algorithm (Nguyen and Madhusudhan, Nucleic Acids Res., 2011) using the main chain atoms Cα as representative atoms. The substructures of two proteins, PDB codes 1xxa chain A (1xxaA) and 1tig chain A (1tigA), are shown in red and blue respectively. The representation of the 3D structure is a composite of the Cα trace, secondary structure, and accessible surface area (shown in mesh representation). The two sets of 6 points encircled in red and blue represent 6-body (n=6) cliques in Clique Matching step. The line that connects them symbolizes the matching of the Cartesian coordinates (by least square fitting), the secondary structure, and the solvent accessible surface area. A pair of n-body cliques is matched if their RMSD on superimposition is smaller than a preset threshold. RMSD between cliques is calculated by 3D least squares fit. Then all matched pairs of n-body cliques are extended to all possible higher order cliques. To begin with, all possible 3-body cliques are compared to one another. Cliques are then extended to a maximum of 7 constituent residues. Matching cliques helps in identifying structurally equivalent residues in the two structures. Using these equivalences, a final 3D least squares fit is performed to superimpose the two structures (Global Alignment step). Given that the matching of cliques is not unique, ie. many cliques comparisons could fit the criteria for a match. Of all the possible least square fits, the comparison that yields the best structure overlap is considered. |
|
|
|
3. Energy minimization step for eliminating target proteins having ugly contacts with the ligand
|
|
|
|
|
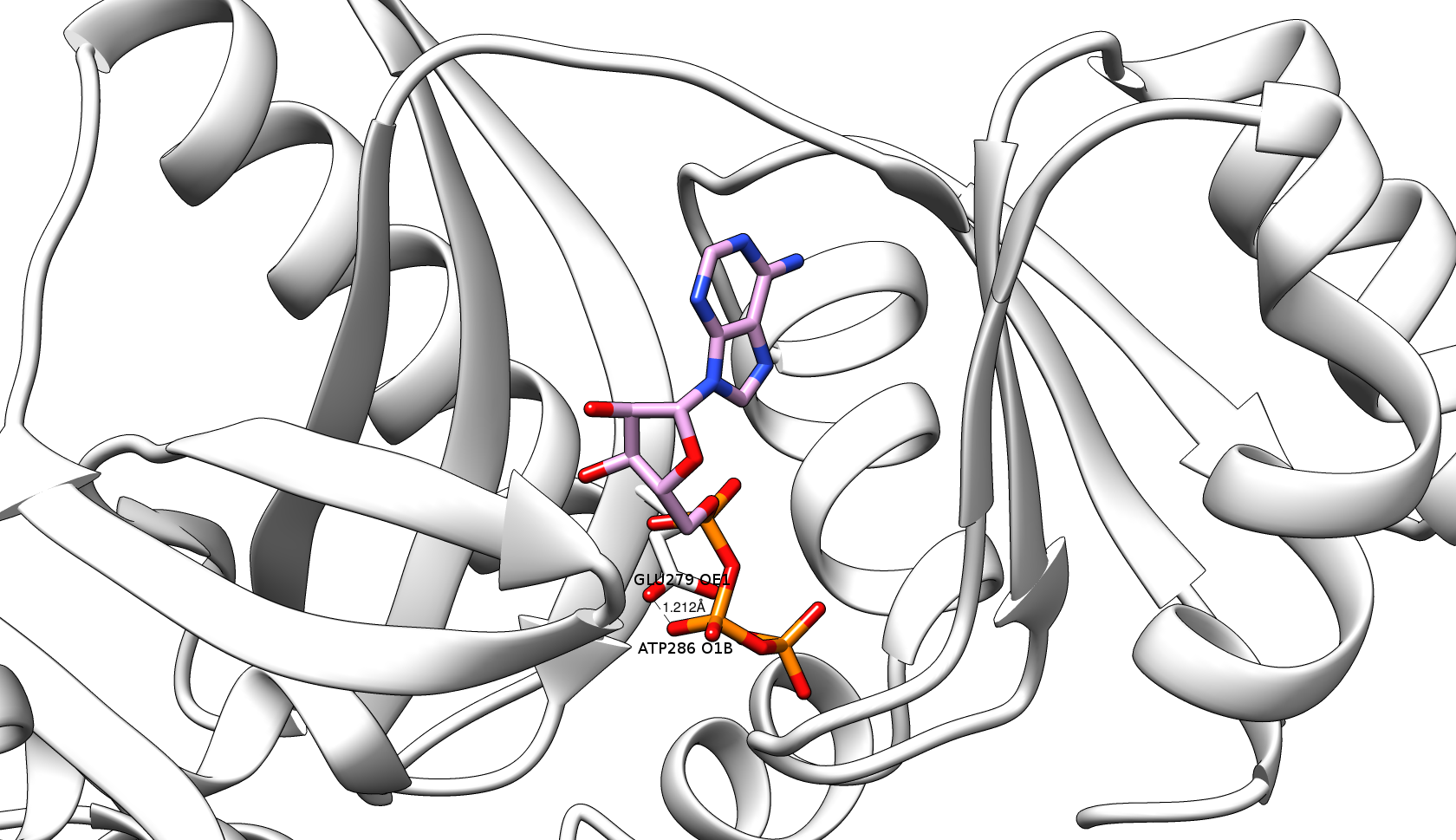
Figure 3: The ugly contact between O1B atom of ATP (Adenosine triphosphate, plum & orange sticks) and OE1 atom of GLU279 (white sticks) of the target protein of PurT-encoded glycinamide ribonucleotide transformylase (gray ribbons) (PDB code: 1kj9A) with the distance = 1.212 Å and Contact score = 0.41 before the energy minimization step. |
|
|
|
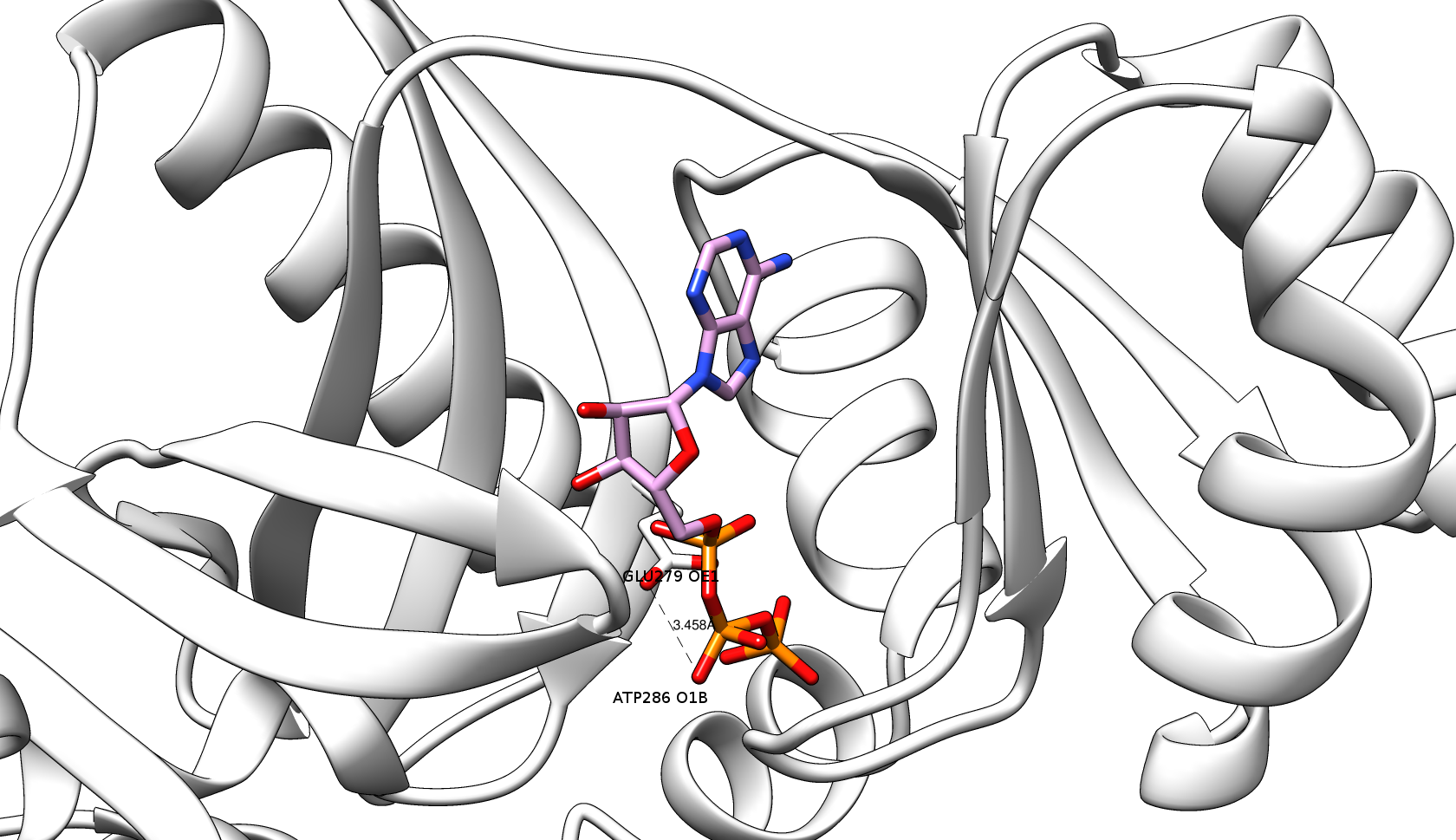
After the virtual screening step , 3Dclick identifies the list of possible target proteins with significant Z-scores (Z-score ≥2). For each of these possible target protein, a complex model is constructed with the ligand in its new putative binding site. To begin with, the complex is simply the coordinates of the ligand transferred onto the new target protein after superimposing the structure of target protein on the binding site of ligand (Figure 1). Then, 3Dclick eliminates proteins that have the significant number of ugly contacts from the list of possible target proteins (as indicated in the virtual screening step ). Here, we would like to have no ugly contact between the non-hydrogen atoms of target proteins and the ligand in the complexes. Therefore, the energy minimization step is applied to attempt removing any possible ugly and bad contacts of the complexes. As shown in Figure 3, there is an ugly contact between O1B atom of ATP and OE1 atom of GLU279 of the target protein of PurT-encoded glycinamide ribonucleotide transformylase (PDB code: 1kj9A) with the distance = 1.212 Å and Contact score = 0.41 before the energy minimization step. For the energy minimization step, 3Dclick first uses AmberTools22 to generate parameter and coordinate files for the ligand using General AMBER Force Field (GAFF2) (He et al., J. Chem. Phys. , 2020). The energy minimization step is skipped if 3Dclick could not generate the parameter and coordinate files for the ligand. After that, a total of 200 steps of energy minimization with the first 50 of steepest descent and the remainder 150 of conjugate gradient is applied using the popular generalized Born (GB) implicit-solvent model of Hawkins et al. (Hawkins et al., J. Phys. Chem. , 1996) with the cutoff of 16 Å for nonbonded pairs (http://ambermd.org/tutorials/basic/tutorial4b/). As shown in Figure 4, the energy minimization step of 3Dclick converts the ugly contact between O1B atom of ATP and OE1 atom of GLU279 of the target protein of PurT-encoded glycinamide ribonucleotide transformylase (PDB code: 1kj9A) (Figure 3) to the good contact with the distance = 3.458 Å and Contact score = 1.17. After the energy minimization step, 3Dclick eliminates any protein in the remaining list of targets if that protein still has ugly contacts with the ligand. Since the energy minimization step usually takes a long running time (for example, the running times of energy minimization step for the targets of PurT-encoded glycinamide ribonucleotide transformylase (PDB code: 1kj9A with 386 residues) and human AlphaFold structure of Obscurin (AF-Q5VST9-F32 with 1400 residues) with ATP using the binding site of A.fulgidus Rio2 Kinase are 4 and 13 minutes, respectively), our 3Dclick web server currently shortlists the top 100 hits/target proteins of significant Z-score and uses them for the energy minimization step. Please connect with us, if users would like to have larger possible target proteins for their ligands/small molecules or drugs. |
|
|
|
|
|
|
Figure 4: Energy minimization step of 3Dclick converts the ugly contact between O1B atom of ATP (plum & orange sticks) and OE1 atom of GLU279 (white sticks) of the target protein of PurT-encoded glycinamide ribonucleotide transformylase (gray ribbons) (PDB code: 1kj9A) to the good contact with the distance = 3.458 Å and Contact score = 1.17.
|