
Analysis, comparison, visualization of contact residues & interfacial waters of antibody-antigen structures/models
|
 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Methods
2. Contact residues 3. Hydrogen bonds 4. Hydrophobic interactions 5. Van der Waals interactions 6. Ionic interactions 7. Interfacial water molecules 8. Root mean square deviation 9. Structure overlap 10. Match size 11. Z-score 1. Graphical representation of AppA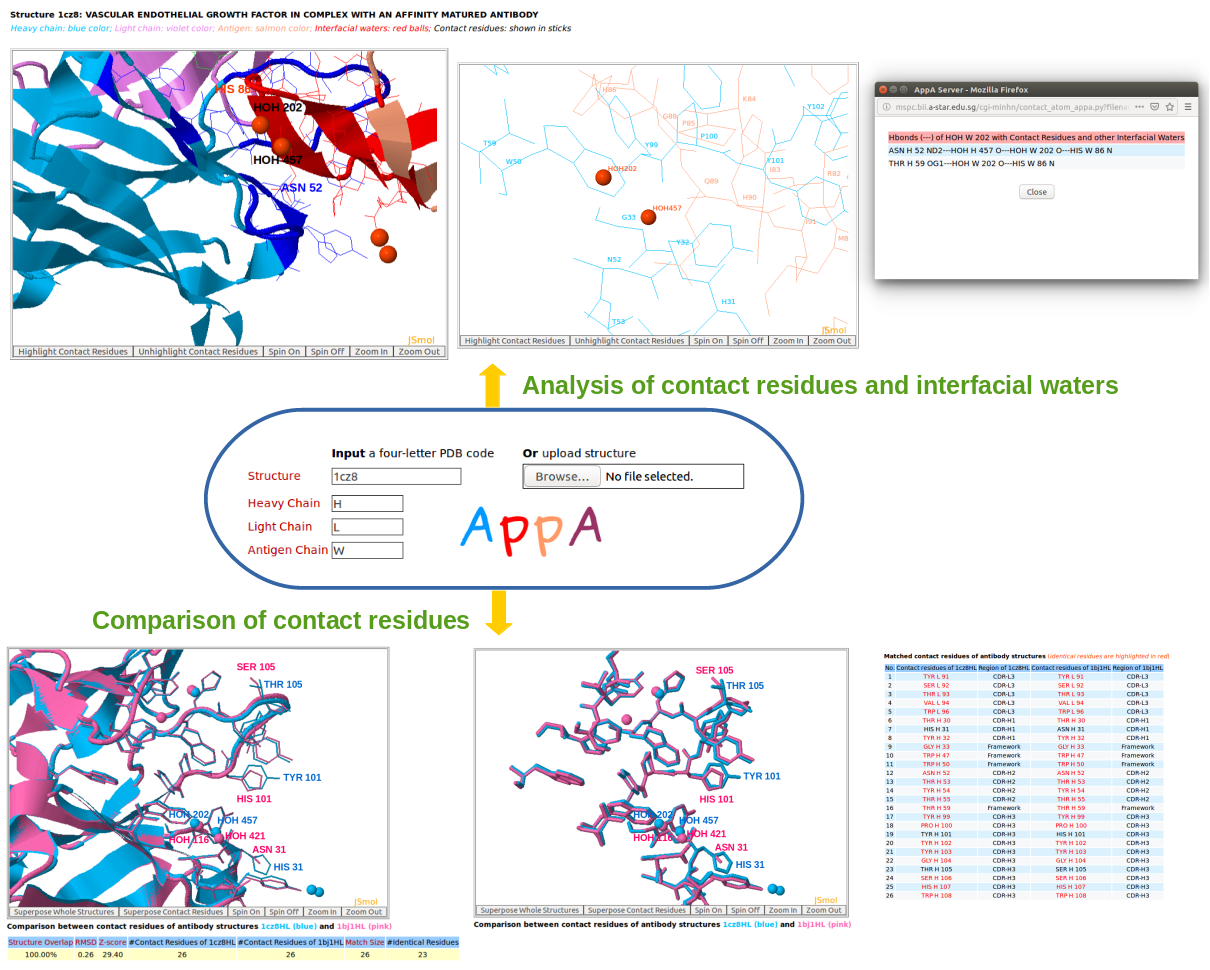
A graphical representation of AppA for analysis and comparison of 3D structures of contact residues and interfacial waters from antibody-antigen structures and the models deposited. Given below are the definitions of contact residues and hydrogen bonds, van der Waals interactions, hydrophobic interactions, and ionic interactions of contact residues and interfacial water molecules that are used to study antibody-antigen interface. AppA has optimized the CLICK algorithm (Nguyen et al., Nucleic Acids Res., 2011) for comparing 3D structures of contact residues of antibody-antigen structures. AppA also calculate Z-score to identify significant comparison/alignment of contact residues of antibody-antigen structures. 2. Contact residuesContact residues are identified as residues of an antibody (antigen) structure whose solvent accessible surface areas (ASA) changes upon the formation of its antibody-antigen complex and they are within 6Å of the complexed epitope (paratope) (Nguyen et al., Bioinformatics, 2017). 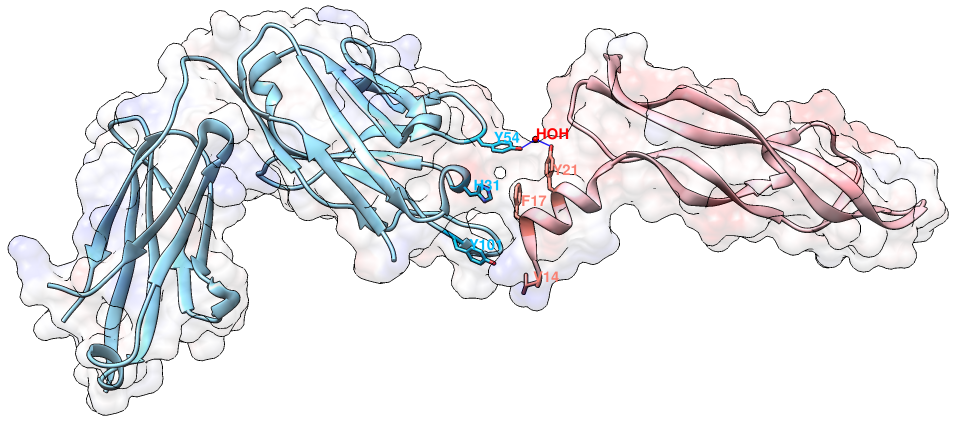
An example of contact residues (blue and salmon sticks) on the heavy chain (H, blue color) of the antibody drug Lucentis (PDB code: 1CZ8) and the antigen VEGF (chain V, salmon color). The solid regions using Chimera show the surface of this complex. The interfacial water HOH509 makes hydrogen bonds with the sidechain of TYR54 of heavy chain of Lucentis and the sidechain of TYR21 of antigen VEGF. The input of this example for AppA: Structure = 1cz8; Heavy Chain = H; Antigen Chain = V. 3. Hydrogen BondsHydrogen bonds are defined for geometries where the angle between the hydrogen bond donor D, the attached hydrogen atom and the hydrogen bond acceptor A ( D–H. . .A) is greater than 120° and the distance between D and A is less than 3.5 Å. The relax hydrogen bond constraints of 0.4 Å and 20 degrees are also applied for AppA to identify hydrogen bonds (Nguyen et al., Bioinformatics, 2017). 4. Hydrophobic interactionsHydrophobic interactions are defined as interactions between any two of the nine non-polar amino acids (Ala, Ile, Leu, Met, Phe, Pro, Val, Tyr, Trp) when their residue distance, measured as a distance between any two non-polar groups in the non-polar sidechains is less than 5Å. 5. Van der Waals interactionsVan der Waals interactions are defined as interactions between any two amino acids when the distance between any two non-hydrogen atoms is less than the sum of the van der Waals radii plus 0.5 Å of the respective atoms. 6. Ionic interactionsIonic interactions are defined as interactions between any two oppositely charged amino acids (Asp, Glu - negatively charged; Lys, Arg, His - positively charged) when the distance between cationic atoms (sidechains NH1, NH2 of Arg; ND1, NE2 of HIS; NZ of Lys) and anionic atoms (sidechains OD1, OD2 of Asp; OE1, OE2 of Glu) is less than 6Å. 7. Interfacial water moleculesWater molecules are identified as interfacial waters if they make hydrogen bonds with both contact residues (or contact water molecules) of the antibody and antigen. Contact waters are water molecules that have direct hydrogen bonds with paratope or epitope (Nguyen et al., Bioinformatics, 2017). 8. Root mean square deviationGiven two 3D structures A and B of contact residues, root mean square deviation (RMSD) is the norm of the distance vector between the two sets of coordinates of representative C-alpha atoms, after superimposition. It is given by 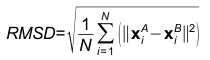
Where, N is the match size, and xiA and xiB are the Cartesian coordinates of representative C-alpha atoms of structurally equivalent residues of structures A and B. 9. Structure overlapStructure overlap (also called equivalent positions) is defined as the percentage of the representative C-alpha atoms in the structure A that are within 2Å (RMSD cut-off) of the corresponding C-alpha atoms in the superimposed structure B. 10. Match sizeMatch size is defined as the number of matched residues in the list of equivalence of structures A and B. 11. Z-scoreFor each comparison/alignment produced by CLICK, a Z-score is computed to determine the significance of the comparison/alignment. The Z-score is an estimate of the likelihood that the alignment is different an alignment made by chance. It is computed as 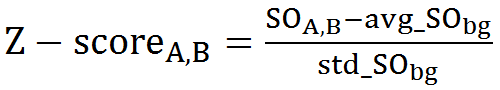
Where, For a significant match, Z-score should be above 2.0 (Nguyen et al., J Chem Inf Model., 2019). The greater the Z-score the more significant is the match. |