Users can choose query structures from the PDB without having to upload their own structure files.
The input can be just the 4 letter PDB code,
in which case the whole structure corresponding to that PDB entry will
be considered. Should you want to specify chains and fragments use the
following format -
- 1ang
The whole structure 1ang
- 1ang,A
The structure 1ang, chain A
- 1xxa,A,B
The structure 1xxa, chains A and B
- 1ang,A(1:15),A(82:100)
The query in this case
would be a 2 fragment structure constituting residues 1-15 and 82-100
of the structure 1ang, chain A.
Note:
The user uploaded structures cannot be fragments like the chosen PDB structures. The user uploaded structures should be pre-fragmented (if so desired).
Representative atoms should be specified exaclty as they appear in the PDB files (upper case, etc).
The choice of representative atoms depends on the types of molecules that are to be aligned.
At the least, 3 representative atoms should be present in both structures to be aligned.
The user can chose more than one type of representative atoms
eg. When aligning a pair of proteins the representative atoms chosen could be "CA CB".
When chosing multiple types of representative atoms separate then with spaces.
Suggested choices of representative atoms
protein-protein alignments: CA; Or any main-chain atom or combination of main chain atoms
DNA/RNA alignments: C3'; Or any nucleotide backbone atom
DNA-protein complexes: CA C3' ; Or any combination of protein/nucleotide backbone atoms
To align ligand molecules change the HETATM label to ATOM
Choose the appropriate representative atom(s).
In addition to the cartesian coordinates of the representative atoms, CLICK can make use of other features of the protein such as secondary structure, depth, etc (see below) in guiding the alignment.
Note:
Secondary structure, side-chain solvent accessibility, and residue depth are currently computed only for protein structures.
Secondary structure provides the general three-dimensional form of local segments of proteins.
For matching of a pair of cliques in our algorithm, the secondary structure score between two equivalent residues Ai and Bj are compared:
SSM[Ai,Bj]< s
Where
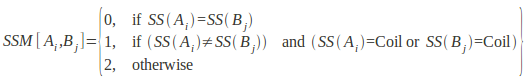
SSM is an empirically determined secondary structure match matrix, SS(Ai) is the secondary structure state of amino acid residue Ai, and s is a preset threshold for matching secondary structure elements. The cut-off threshold for comparing secondary structure used in this study was 2, hence SSM[Ai,Bj]< 2. This implies that, either residues of regular secondary structures can only match with other residues of the same secondary structure, or with residues in loops.
Side-chain solvent accessibility is the degree to which a residue in a protein is accessible to a solvent molecule.
The solvent accessibility score between two residues Ai and Bj are matched by using the inequality:
SAM[Ai,Bj]< a
Where
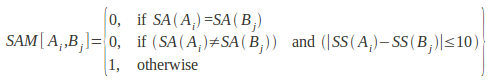
SAM is an empirical solvent accessibility match matrix, SA(Ai) is the side-chain solvent accessibility of amino acid residue Ai, and a is a preset threshold for matching solvent accessible area states. The cut-off threshold for solvent accessibility matching is a=1, implying that residues categorized in different accessible area classes cannot be matched. However, this criterion is relaxed to allow the matching of two residues in adjacent accessible area classes if their side chain accessible areas are within 10% of each other.
Depth measures the closest distance of a residue/atom to bulk solvent. A detailed description of the computation of depth can be found here.
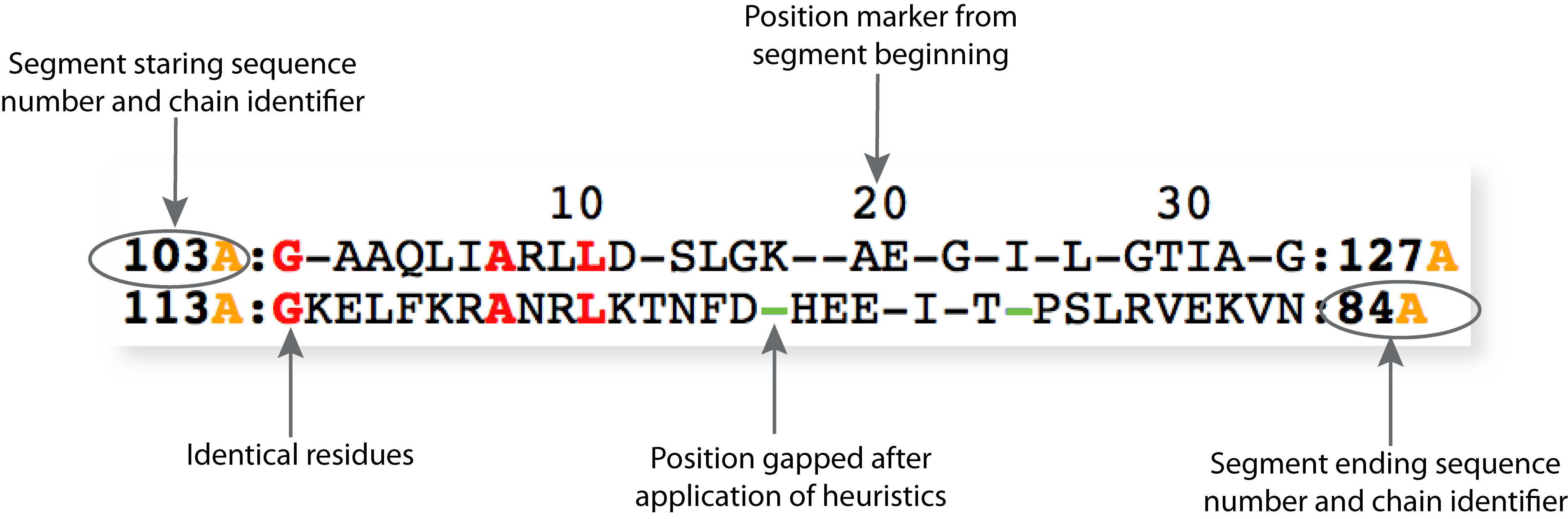
Heurisitics are applied to ensure local chain connectivity.
Chain connectivity is broken when equivalent residues are not contiguous in sequence ( eg. residue highlighted in the figure below).

The equivalent residue matches before the application of heuristics is shown in the output file linked to "Matched residue pairs".
Rules:
Matched segments must have a minimum length of 5 residues (gap matching not included).
Residue matches that do not follow this criteria are removed
Adjacent segments separated by 5 or less residues are joined together.
Currently, users can search over structural databases of
non-redundant (90% sequence identity) set of 22,834 protein chains
non-redundant set of 4239 human protein chains
non-redundant set of 1368 DNA
a set of 2262 DNA-protein complexes
non-redundant set of 493 RNA
The server offers the users the flexibility of uploading their own database, should the default ones be inadequate for their queries. The format of users' database is as follows: the first line is the type of their database (protein/DNA/RNA or DNA-protein) and next lines are just the letter PDB code and/or chain - eg.
protein
7odcA
4eetB
3mhpC
3c4s
1xak
3l32
As databases could be large, you may want to restrict the number of
comparisons for which you may require detailed information (alignment
files and the superimposed structures). The hits are ranked
according to Z-scores.
This is an optional field. A link to the results page will be sent to the email address provided.
For jobs that could take a long time (large databases), it may be useful to provide an e-mail id.
If no address is provided care should be taken to save the URL that points to the results immediately after submitting a job. This URL refreshes every minute and display the results as soon as the job terminates.
The running time increases with increase in
size of the query structure
size of database
number of representative atoms
number of best matched cliques for each alignment
Below table lists running times of query structures with different lengths using the non-redundant dataset of 24,563 protein chains and CA representative atom.
Note that these are estimated running times. Jobs may also queue up on the server.
| Length of query structures (no. of residues) |
100 |
200 |
300 |
500 |
| Running time (hours) |
~4 |
~6 |
~10 |
~24 |
|
